
La fibrosis quística es una enfermedad genética y hereditaria que afecta a la fisiología de los pulmones debido a que las mucosas son demasiado espesas. Veamos sus características clínicas.

Pol Bertran Prieto
Microbiólogo, divulgador científico y Youtuber
Temas relacionados
Genética
Enfermedades
Pulmones
Los seres humanos somos el resultado de la interacción entre nuestros 30.000 genes y el ambiente. Y estos genes no son, por desgracia, unidades indestructibles. Estos segmentos de ADN que codifican para proteínas pueden presentar errores en su secuencia de nucleótidos que llevan a que ciertas células no puedan cumplir con sus funciones fisiológicas.
Cuando la persona presenta un error genético, es posible que desarrolle lo que se conoce como enfermedad genética, la cual, si viene acompañada de una herencia a la descendencia a través de los gametos sexuales, es también una enfermedad hereditaria.
Se cree que, debido a la gran variedad de genes y a la aleatoriedad en las mutaciones, podrían haber más de 6.000 enfermedades genéticas, pero es evidente que hay algunas que presentan una incidencia mayor que otras. Y este es el caso de la fibrosis quística, una patología genética y hereditaria con una incidencia de 1 caso por cada 3.000-8.000 personas.
Así pues, en el artículo de hoy y de la mano de las más recientes y prestigiosas publicaciones científicas, te traemos una selección de la información clínica más relevante acerca de la fibrosis quística, una enfermedad que afecta a la fisiología de los pulmones, así como a la del aparato digestivo y de otros órganos del cuerpo. Empecemos.
Qué es la fibrosis quística?
La fibrosis quística es una enfermedad genética y hereditaria potencialmente mortal que consiste en la acumulación de mucosidad inusualmente espesas y pegajosas en los pulmones, tracto digestivo y otras regiones del cuerpo. Es una de las formas de patología pulmonar crónica más común en niños y adultos jóvenes.
Se trata de un trastorno heredado que causa daños graves en la fisiología principalmente pulmonar y digestiva, pues los errores genéticos se manifiestan con una alteración de la funcionalidad de las células que producen moco, jugos digestivos y sudor. La afectación a un gen hace que no produzcan líquidos ligeros y resbaladizos, sino espesos y pegajosos.
Estas secreciones, en lugar de cumplir con su función de lubricación en los órganos correspondientes, se acumulan y tapan los tubos y conductos principalmente de los pulmones y del páncreas, un órgano de naturaleza glandular que forma parte del sistema tanto digestivo (libera enzimas digestivas) como endocrino (libera hormonas que regulan los niveles de glucosa).
La falta de aliento, la tos persistente, las obstrucciones intestinales, el sudor muy salado, la tendencia a sufrir infecciones pulmonares, la congestión nasal, los retrasos en el crecimiento, la mucosidad constante, etc, son los principales síntomas de una enfermedad que, con el tiempo, va empeorando.
Esta enfermedad no tiene cura al tratarse de un trastorno genético y, a pesar de que gracias al diagnóstico precoz (suele detectarse entre el primer mes y los 2 años de edad) y a la aplicación de cuidados para controlar su progreso se ha mejorado la calidad y esperanza de vida de los afectados, las personas con fibrosis quística viven hasta los 30, 40 o, en algunos casos, los 50 años. Las infecciones pulmonares y los graves problemas digestivos explican esta mortalidad.
Causas
Las causas de la fibrosis quística están muy bien descritas. Como hemos dicho, se trata de una enfermedad genética y hereditaria, por lo que su aparición se debe a errores en la secuencia de un gen que se heredan de padres a hijos. Sea como sea, cabe recalcar que su incidencia es de 1 caso por cada 3.000-8.000 nacimientos vivos.
Pero, cuál es el error genético que da lugar a la fibrosis quística? La fibrosis quística surge por una mutación en el gen CFTR, el cual se encuentra en el cromosoma 7 (locus 7q31.2), un gen que codifica para la proteína reguladora de la conductancia transmembrana de la fibrosis quística.
En condiciones normales, el gen CFTR codifica para unas proteínas que controlan el paso de los iones cloro a través de las membranas celulares de las células productoras de líquido para así asegurar que estos sean ligeros y resbaladizos.
Por desgracia, hay más de 1.500 defectos (mutaciones) genéticos que pueden derivar en una deficiencia en este gen, lo que impide que la persona produzca estas proteínas, cosa que, a su vez, causará que las mucosidades sean más pegajosas de lo normal. Dependiendo de la mutación específica, la gravedad de la fibrosis quística será mayor o menor.
Y, ¿cómo se heredan estas mutaciones? Las mutaciones en el gen CFTR siguen un patrón de herencia autosómica recesiva. Nos explicamos. Los seres humanos tenemos 23 pares de cromosomas, es decir, dos copias de cada cromosoma. Por lo tanto, como hay dos copias del cromosoma 7, tenemos también dos copias del gen CFTR.
Y como el patrón es recesivo, si solo uno de los dos genes CFTR es defectuoso (está mutado), no pasará absolutamente nada. Habrá la otra copia buena para compensar. Un gen estará mutado, pero el otro permitirá que se siga sintetizando la proteína que hemos comentado.
En este sentido, una persona solo desarrolla fibrosis quística cuando dispone de los dos genes CFTR mutados. Ha tenido que recibir ambos genes mutados de los dos padres. Es decir, si el padre es portador de la mutación (tiene solo un gen mutado, así que no expresa la enfermedad) pero la madre no es ni siquiera portadora, el riesgo de que alguno de sus hijos desarrolle fibrosis quística es, pese a que el padre porte la mutación, de 0%.
Pero si tanto el padre como la parte son portadores (ninguno sufre la enfermedad pero ambos tienen una de las dos copias mutadas), el riesgo de que alguno de sus hijos herede los dos genes (y, por tanto, desarrolle la enfermedad) es del 25%. En esto se basa la herencia recesiva.
Y esto también explica que, pese a que la incidencia sea, de media, de 1 caso por cada 5.000 nacimientos vivos, se calcule que 1 de cada 25 personas son portadoras del gen CFTR mutado. Nunca expresarán la enfermedad pero, en caso de tener descendencia con otra portadora, harán que haya riesgo de que sus hijos padezcan la fibrosis quística.
Más allá de esto, cabe recalcar también que la enfermedad es más común en personas caucásicas (especialmente del centro y norte de Europa). Aun así, además de, evidentemente, antecedentes familiares de la patología, no se conocen más factores de riesgo asociados.
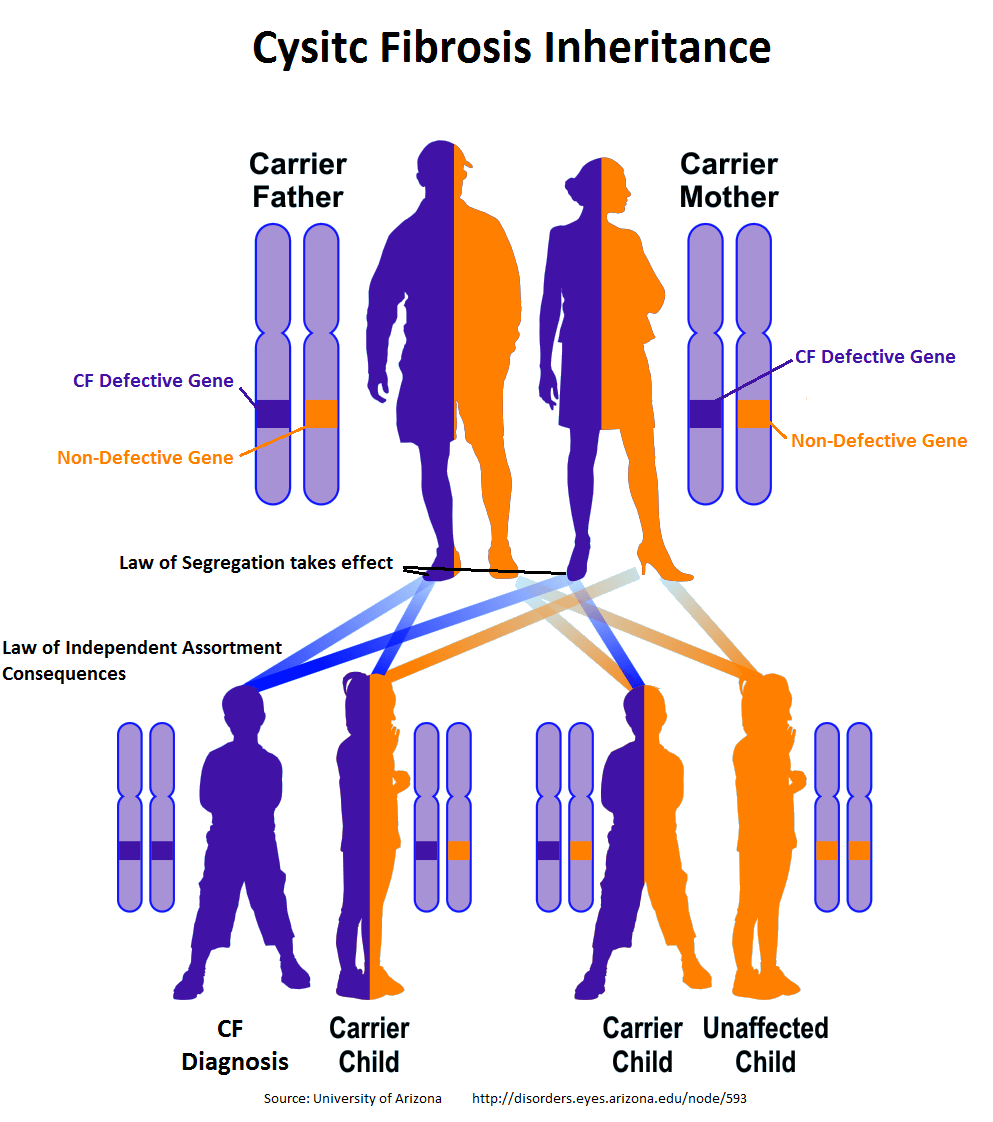
La herencia de la fibrosis quística sigue un patrón recesivo.
Síntomas
Como hemos dicho, hay más de 1.500 mutaciones en el gen CFTR que pueden derivar en el desarrollo de la fibrosis quística. Por lo tanto, las manifestaciones clínicas, su momento de aparición y su gravedad variarán de persona a persona.
Sea como sea, los síntomas respiratorios más comunes aparecen por la acumulación de moco en los pulmones y suelen consistir en: sibilancia (silbidos al respirar), tos persistente, esputo (mocos espesos), intolerancia al ejercicio, congestión nasal, inflamación de las fosas nasales, sinusitis recurrentes y tendencia a sufrir infecciones pulmonares.
Por otro lado, surgen también síntomas digestivos debidos principalmente al bloqueo de los conductos pancreáticos a causa del moco espeso (el páncreas no puede liberar sus enzimas digestivas al intestino delgado) y que consisten en: estreñimiento, prolapsos rectales, heces grasosas, heces de muy mal olor, problemas para subir de peso, obstrucciones intestinales, pérdida de apetito y náuseas
Paralelamente, también son habituales los retrasos en el crecimiento (derivados de los problemas digestivos), que el sudor sea inusualmente salado y la fatiga. Pero el verdadero problema es que, con el tiempo, la fibrosis quística deriva en complicaciones más graves.
Infecciones pulmonares crónicas, daños a las vías respiratorias, tos con sangre, pólipos nasales, neumotórax (el aire se filtra al espacio que separa los pulmones de la pared torácica, provocando el colapso de parte o la totalidad del pulmón), insuficiencias respiratorias, deficiencias nutricionales, diabetes de tipo 2 (hasta el 50% de los adultos desarrollan diabetes ya que el páncreas no puede producir niveles óptimos de insulina), pancreatitis, enfermedades hepáticas, osteoporosis, problemas de salud mental, deshidratación, reducción de la fertilidad en mujeres e infertilidad en hombres. Estas son las principales complicaciones.
Todo ello explica que, pese a que los tratamientos que ahora comentaremos hayan supuesto una enorme mejora en la calidad y esperanza de vida de las personas con fibrosis quística, las personas afectadas por esta enfermedad vivan, de media, 35 años. Aun así, en países con sistemas de salud más avanzados (y dependiendo de la gravedad de la patología), la esperanza de vida puede llegar a los 50 años. Las infecciones pulmonares y obstrucciones bronquiales están detrás del 95% de fallecimientos en personas con fibrosis quística.
Tratamiento
La fibrosis quística es una enfermedad genética y hereditaria, por lo que no es ni prevenible (a no ser que la pareja se haga pruebas genéticas) ni curable. Aun así, se han desarrollado opciones de tratamiento tanto para aumentar la calidad de vida de los pacientes como para incrementar su esperanza de vida.
El diagnóstico se basa en exámenes rutinarios en los recién nacidos, donde, a través de un examen de sangre, se miden los niveles de tripsinógeno inmunorreactivo, una sustancia producida por el páncreas que, en caso de ser alto, es un indicador de posible caso de fibrosis quística. En caso de que haya sospechas, se realizará un test de sudor, donde se comprueba si el sudor es más salado de lo normal. Y si siguen habiendo sospechas, se realizará una prueba genética para confirmar o rechazar el diagnóstico.
Hay que tener claro que, tras un diagnóstico positivo, empezará un control muy estricto y una intervención tanto temprana como agresiva para ralentizar al máximo el progreso de la enfermedad, prevenir y controlar las infecciones pulmonares, garantizar una correcta nutrición, prevenir las obstrucciones intestinales y extraer la mucosidad acumulada en los pulmones.
El tratamiento consistirá en la administración de medicamentos antiinflamatorios, antibióticos, ablandadores de heces, enzimas pancreáticas (para contrarrestar la falta de las naturales), reductores de ácido estomacal, broncodilatadores, diluyentes de la mucosidad… Dependiendo de las necesidades.
Existen también unos nuevos medicamentos que actúan como moduladores de la proteína reguladora de la conductancia transmembrana de la fibrosis quística, mejorando su funcionamiento (contrarrestando la mutación en el gen CFTR) y reduciendo los daños pulmonares.
Paralelamente, las sesiones de fisioterapia torácica, mediante técnicas de despeje de las vías respiratorias, pueden aliviar las obstrucciones, rebajar la inflamación de las vías respiratorias y reducir el riesgo de infecciones pulmonares, aflojar las mucosidad y aliviar la tos. Del mismo modo, es posible que los médicos recomienden programas de rehabilitación pulmonar.
Más allá de esto, es evidente que pueden darse tratamientos para abordar las complicaciones de la fibrosis quística, tales como cirugías nasales (si se han desarrollado pólipos que dificultan la respiración), sonda de alimentación, trasplante de pulmón, cirugía intestinal, trasplante de hígado u oxigenoterapia (si los niveles de oxígeno en sangre disminuyen). Gracias a todo esto, pese a que es inevitable que la esperanza de vida se reduzca, poco a poco vamos avanzando en el tratamiento de una enfermedad que, por desgracia, seguirá siendo incurable.





Post comments